
I am a Genentech Endowed tenure-track assistant professor at UW Biostatistics, and started in Fall 2023. Previously, I received my Ph.D. from the Department of Statistics & Data Science at Carnegie Mellon University, under supervision of Dr. Kathryn Roeder and Dr. Jing Lei, and completed my post-doctoral training with Dr. Nancy R. Zhang.
My research focuses on human cellular mechanisms in Alzheimer’s disease (AD) using single-cell data from post-mortem tissue (sequencing and imaging). Statistical methods offer powerful clues, but these current methods leave many fundamental questions unanswered. • (1) How can we use post-mortem tissue to infer deteriorating cellular mechanisms ante-mortem? We can detect differences between tissue from AD donors and cognitively/pathologically normal donors, but do these differences reflect long-term dynamics that persisted for decades before death? • (2) Brain cells respond not only to amyloid-β and neurofibrillary tau tangles (the defining lesions of AD) but these proteins spread differently across the brain, often influenced by co-occurring neurodegenerative diseases. How can we isolate AD-specific cellular responses amid substantial person-to-person variation, region-specific progression, aging, and unmeasured co-pathologies? • (3) Some donors show high AD pathological burden post-mortem yet no ante-mortem cognitive decline. How can we rigorously quantify this "resilience," and what protective mechanisms allow these donors to mitigate the neurotoxicity of amyloid-β and tau? To address all these different questions about AD, my lab develops statistical and computational advances (ex: deep learning, matrix factorization, and assumption-lean hypothesis testing) to build the next generation of dry-lab tools to inform future therapeutic strategies.
Funding: It is my honor to be funded by the NIGMS R35 (2026-31) and the UW ADRC (2026-27) to support the lab. See our lab's past funding/awards.
Course notes: With help from student volunteers, I also maintain online lecture notes that stemmed from "BIOST 545: Biostatistical Methods for Big Omics Data" (Winter 2025), which provides an overview of how statistical/computational methods specialize to understand different 'omics from single-cell sequencing data.
Note: There are multiple "Kevin Lin"'s at the University of Washington. Be sure to contact the correct email (ID: kzlin)!
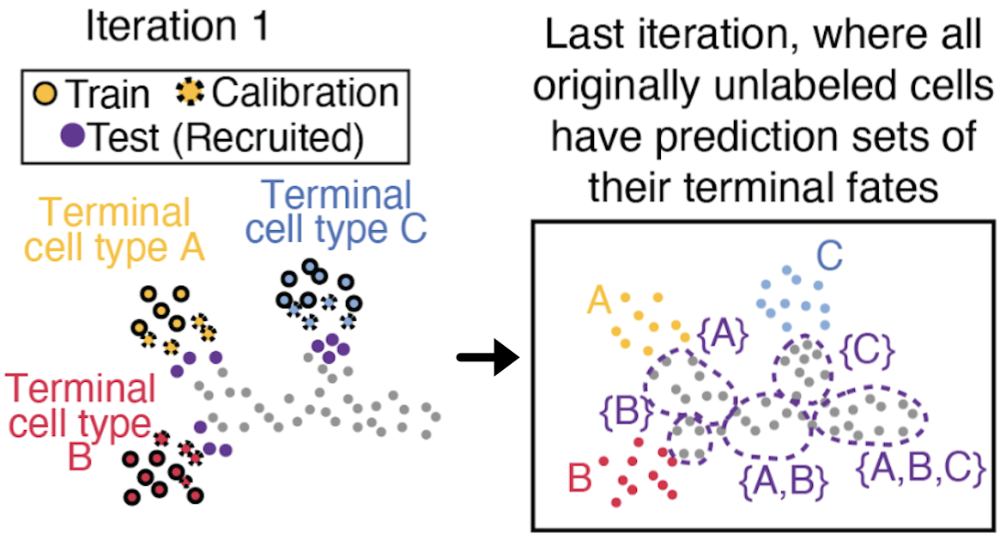
Defining the discrete boundaries of lineage commitment and the timing of regulatory priming remains a significant challenge in developmental biology. We present SCOPE (Semi-supervised Conformal Prediction), a framework that leverages conformal inference to transform high-dimensional single-cell measurements into rigorous, discrete prediction sets of all plausible future fates. By formalizing fate uncertainty with distribution-free coverage guarantees, SCOPE localizes transcriptomic branchpoints and identifies windows of epigenetic priming where chromatin accessibility foreshadows gene expression. We demonstrate its utility across mouse hematopoiesis and human retinogenesis, identifying key transcription factor drivers that define the onset of lineage specification.
SCOPE: Localizing fate-decision states and their regulatory drivers in single-cell differentiation
2026: (biorxiv) (code: method and analysis)
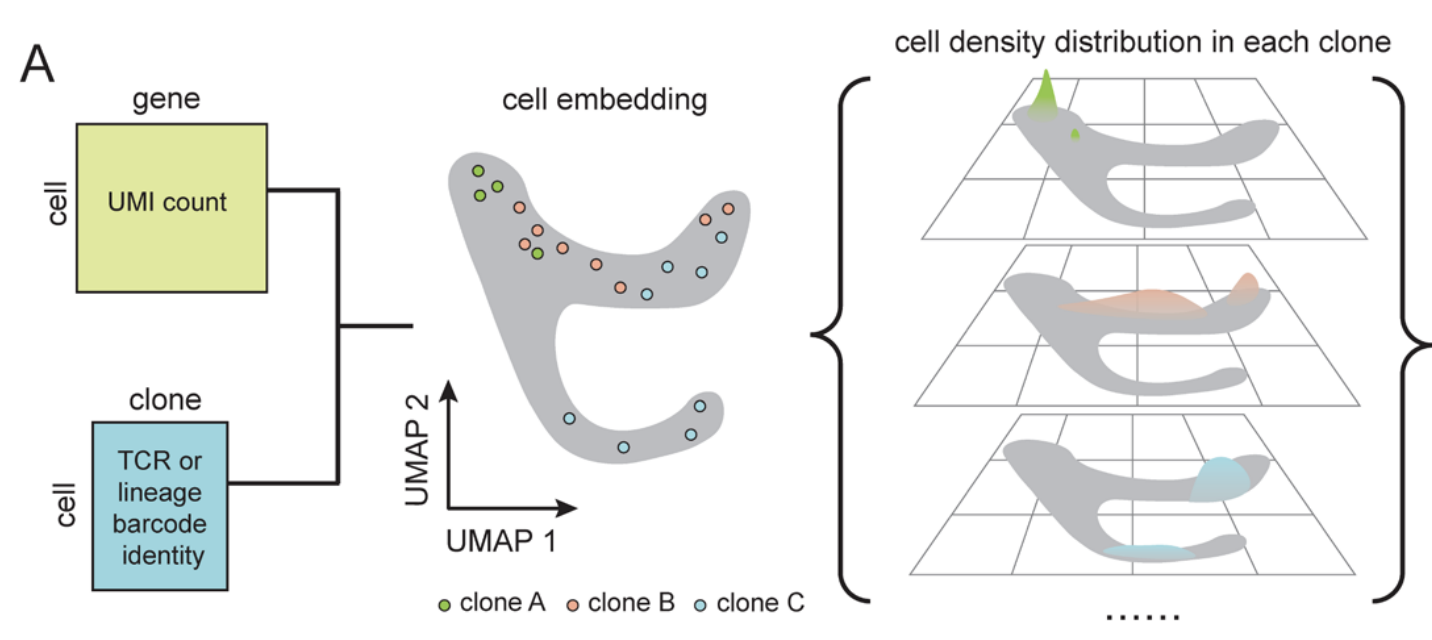
Single-cell technologies now measure both gene expression and clonal ancestry, but most analysis tools still treat these modalities separately. We develop a framework that clusters clones by their "clonotype profiles" (the distributions of their cells across transcriptomic space) to jointly model lineage and expression and recover cell-state transitions and fate biases. Applied to simulated and experimental systems including hematopoiesis, cancer drug resistance models, and CD8 T cells from glioblastoma and immunotherapy cohorts, our method reconstructs temporal order across abrupt transcriptional shifts, distinguishes unipotent from multipotent progenitors, and uncovers subtle, recurrent fate-divergent T cell programs.
Deciphering Cell Fate and Clonal Dynamics via Integrative Single-Cell Lineage Modeling
2025: (biorxiv) (code: method)
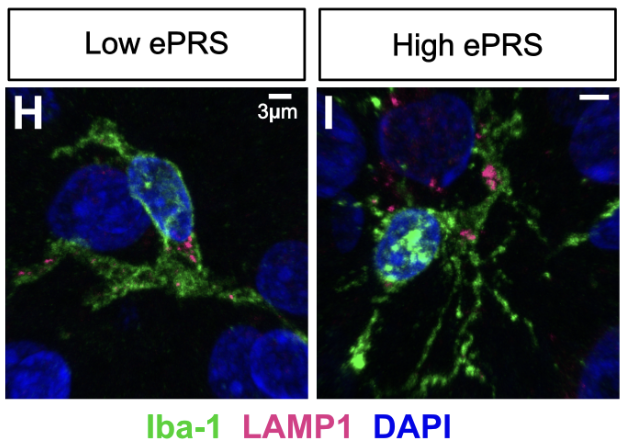
It is unclear whether how genetic variants linked to Alzheimer’s disease (AD) like the endolysosomal network (ELN) cause functional disruptions in the brain. We developed a pathway-specific polygenic risk score (ePRS) based on 13 ELN-related AD GWAS loci and analyzed single-nucleus transcriptomes and endolysosomal morphology in post-mortem DLPFC samples from AD patients stratified by ePRS burden. Using confocal imaging and snRNA-seq, we identified cell-type-specific morphological and transcriptional signatures associated with higher ELN genetic risk, including altered endosome and lysosome structures, dysregulation of glutamatergic signaling, and increased markers of cellular stress. These findings demonstrate that genetic risk in ELN genes contributes to distinct and shared cellular phenotypes across neural cell types, suggesting that pathway-specific risk variants may drive mechanistic heterogeneity in AD.
Genetic risk in endolysosomal network genes correlates with endolysosomal dysfunction across neural cell types in Alzheimer's disease
2025: (biorxiv)
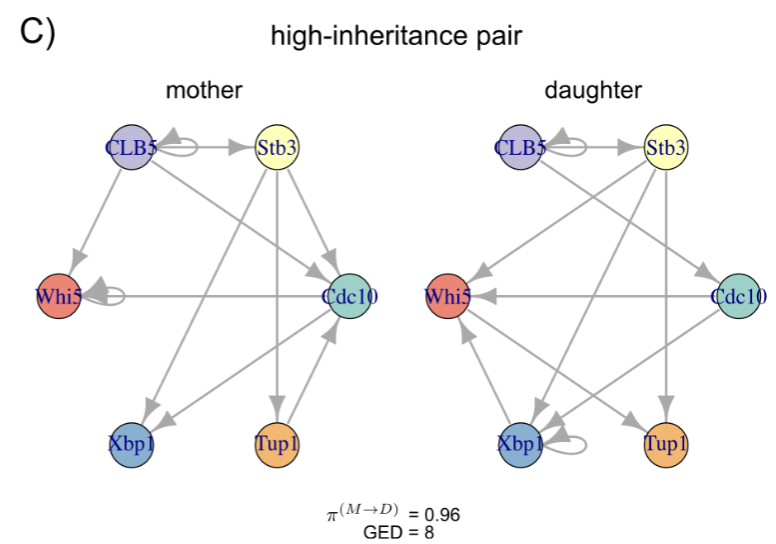
Cells can undergo asymmetric cell division, and in certain systems, the fate of the daughter cell can be predicted even before it buds from the mother cell. However, it is not well understood what exactly is inherited by the daughter cell. We study budding yeast cells via live-cell microscopy to study the protein-protein regulatory network of mother cells and their daughter cells as time series, and develop a framework based on ODE regulatory networks to estimate the the precentage a daughter cell inherits its protein-protein regulatory network from its mother at single-cell resolution.
Measuring regulatory network inheritance in dividing yeast cells using ordinary differential equations
2024: (biorxiv) (code: method) (code: tutorial)
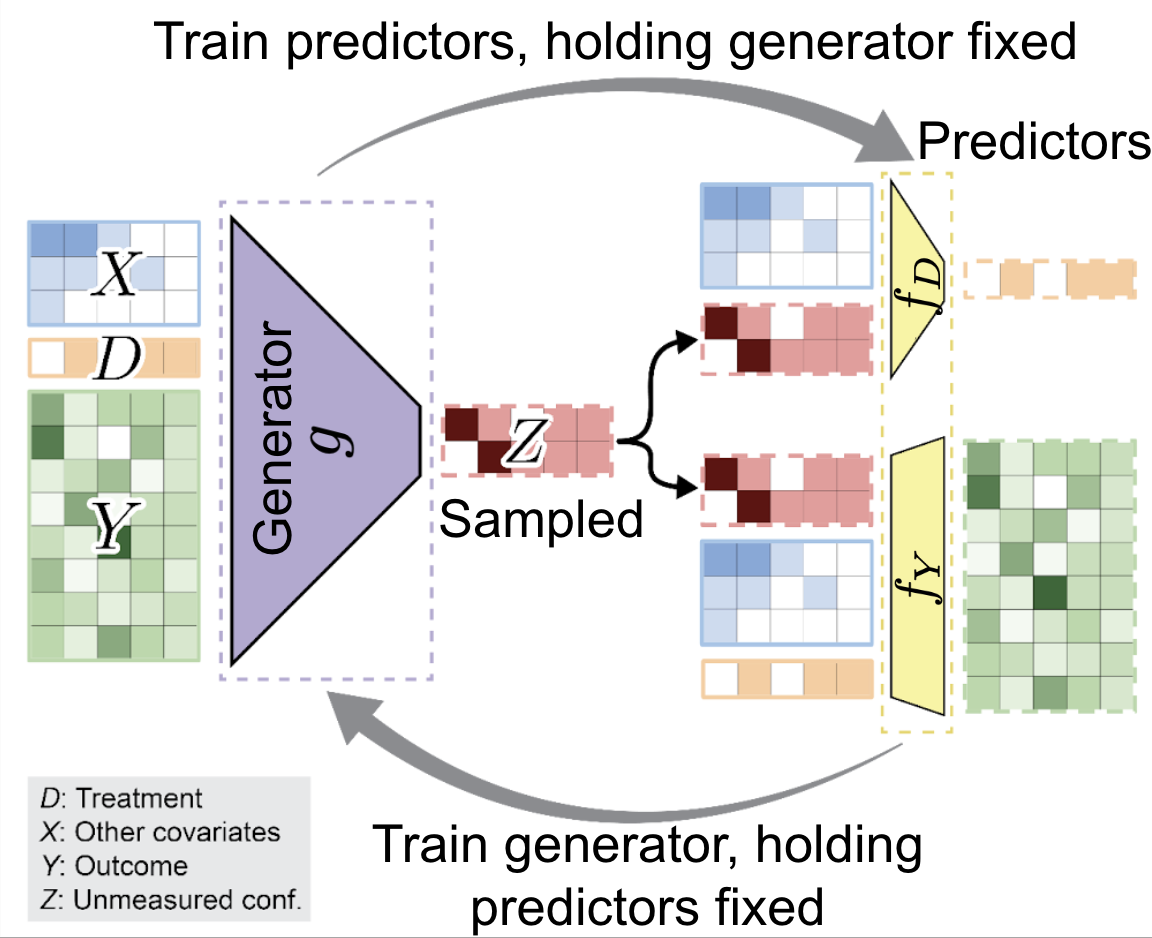
High-dimensional genomics studies are frequently confounded by unmeasured biological processes that obscure disease-specific signals. We introduce sensGAN, a deep-learning adversarial framework that systematically explores the confounding spectrum by learning "worst-case" latent variables that nullify gene associations under novel predictive-gain constraints. By treating confounding as a spectrum rather than a fixed correction, we can distinguish between transcripts robust to latent biological processes and those likely to be artifacts of omitted variable bias. We demonstrate that sensGAN outperforms existing surrogate-variable methods in simulations and successfully prioritizes robust disease pathways in human Alzheimer's disease microglia.
High-Dimensional Sensitivity Analysis for Genomic Studies: An Adversarial Framework for Learning Worst-Case Latent Confounders
International Conference of Machine Learning: To be published 2026 (preprint main) (preprint appendix) (code: method and analysis)
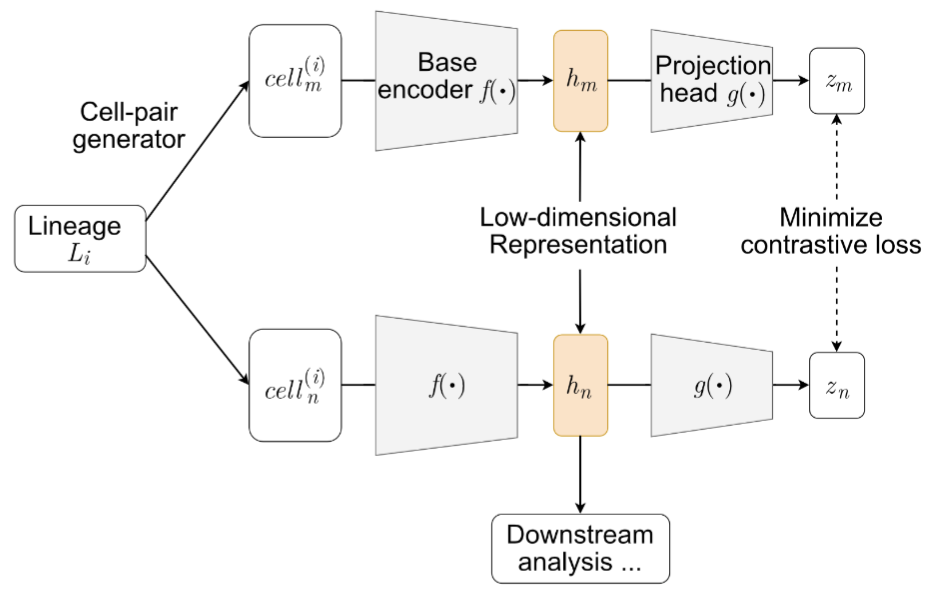
In static lineage-barcoding single-cell sequencing, we measure both each cell's gene expression and its ancestry in order to gain insights into a cell's fate. However, there lack methods to identify which genes separate the cells by their lineages. In this work, we develop a deep-learning architecture based on contrastive learning to isolate the information in the gene expression that recapitulates cells' lineages. We use the lineage barcoe as the premise of our data augmentation mechanism. This strategy enables us to better identify the genes driving fate commitment and cells' fate boundaries.
Leveraging lineage barcodes as natural augmentations for contrastive learning of cell fate in scRNA-seq data
International Conference of Machine Learning: To be published 2026 (preprint main) (preprint appendix) (biorxiv) (code: method and analysis)
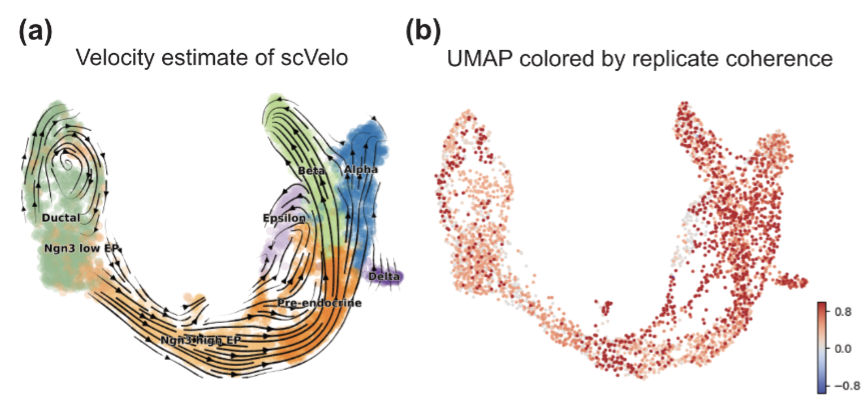
RNA velocity has been a transformative tool to study development biology via single-cell RNA sequencing. However, although many RNA velocity methods exist, it is common for many methods to conflict in the biological interpretation of the results. We develop a method called replicate coherence to quantify the uncertainty in the RNA velocity vectors where we use the count-splitting framework to emulate having two technical replicates of the same exact cells. This allows us to quantify how much uncertainty each RNA velocity result has, which enables a more accurate side-by-side comparison of different methods for the scRNA-seq dataset at hand.
Quantifying stability via count splitting to guide model selection in RNA velocity analyses
Bioinformatics Advances: To be published (biorxiv) (code: method and analysis)
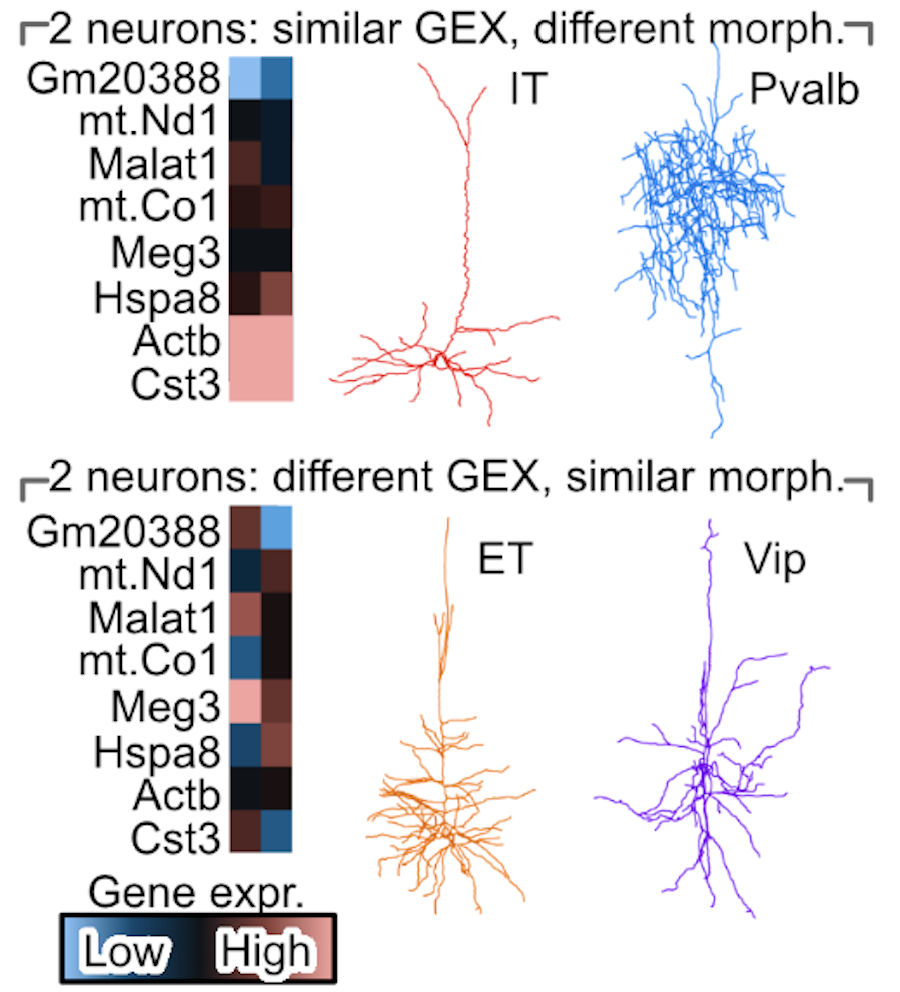
A cell's morphology is a powerful readout of a cell's function and state. While microscopy allows us to study neural cells' morphology, and single-nuclei RNA-sequencing allows us to study a cell's transcriptomics, these two rich data sources are often performed on separate cells due to technological limitations. This obstructs us from understanding how a neural cell's transcriptomics modulates or responds to changes in its morphology. We develop a deep-learning diagonal integration method, to overcome this technological limitation. We validate our method on Patch-seq neurons and study genes that control microglial morphology from the 5xFAD mouse model for Alzheimer's disease.
Integrating morphology and gene expression of neural cells in unpaired single-cell data using GeoAdvAE
RECOMB Proceedings 2026: (link) (code: method and analysis)
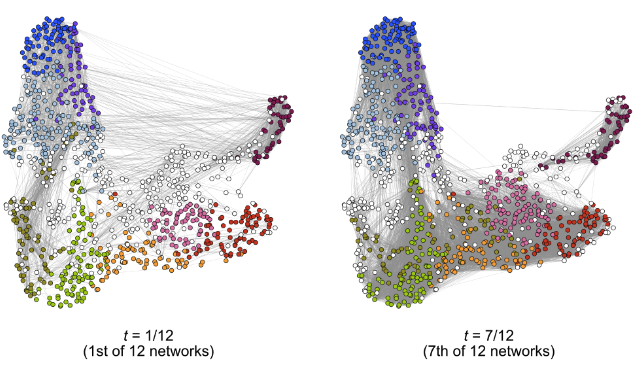
In genomics, we often observe a sequence of graphs over some notion of time, such as the gene network ordered by the cells' developmental age. However, there is a lack of concrete statistical theory for this setting whose assumptions are amendable for single-cell analyses. In this work, we develop an estimator designed for models where the underlying stochastic block model is smoothly changing with time, and prove the convergence rates of the clustering and estimates of the connectivity matrix under assumptions more general than those existing in the literature. We apply this to study the dynamics of gene co-expression networks among oligodendrocytes.
Dynamic clustering for heterophilic stochastic block models with time-varying node memberships
Biometrika: To be published (2026) (link) (arxiv) (code: method and analysis)
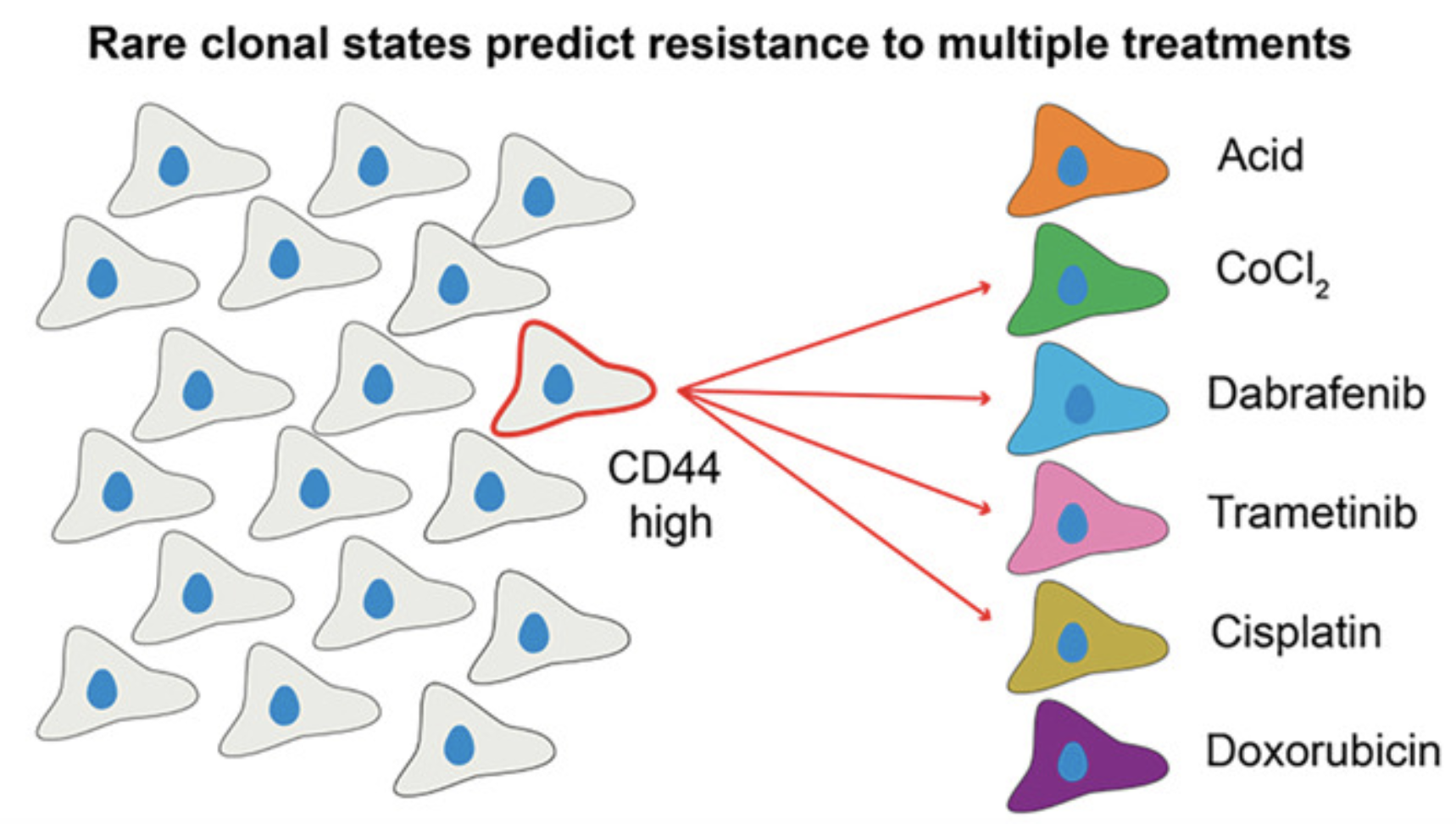
While resistance to single cancer treatments is well-documented, clinical failure often involves multiple therapeutic modalities. We developed a multi-treatment, high-throughput clonal tracking framework paired with single-cell RNA sequencing to monitor melanoma clones across parallel treatment conditions. We discovered rare "generalist" clones that survive multiple diverse treatments and identified pre-treatment CD44 expression as a marker for multi-treatment resistance. Our results demonstrate that initial differentiation states drive divergent transcriptional paths toward resistance, providing a framework for mapping how pre-existing cellular heterogeneity dictates complex therapeutic outcomes.
Pre-existing cell states predict resistance to multiple treatments
Cell Genomics 6 (2026): To be published (link) (code)
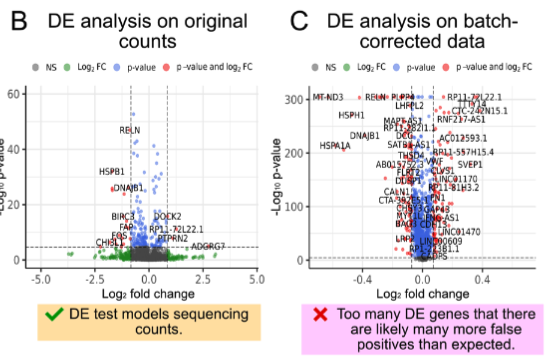
Computational single-cell analyses have become quite complex. While there are many tools for any particular specific analysis, researchers new to single-cell analyses might often feel overwhelmed and not immediately understand the logical flow that connects one computational method to another. This review paper highlights the underlying logic of how to craft a computational workflow, tailored for the biological context of studying glial cells. It starts with thinking about the experimental design, and spotlights common pitfalls when planning your own computational workflow.
All the single cells: Single-cell transcriptomics/epigenomics experimental design and analysis considerations for glial biologists
Glia 73 (2025): (link) (arxiv)
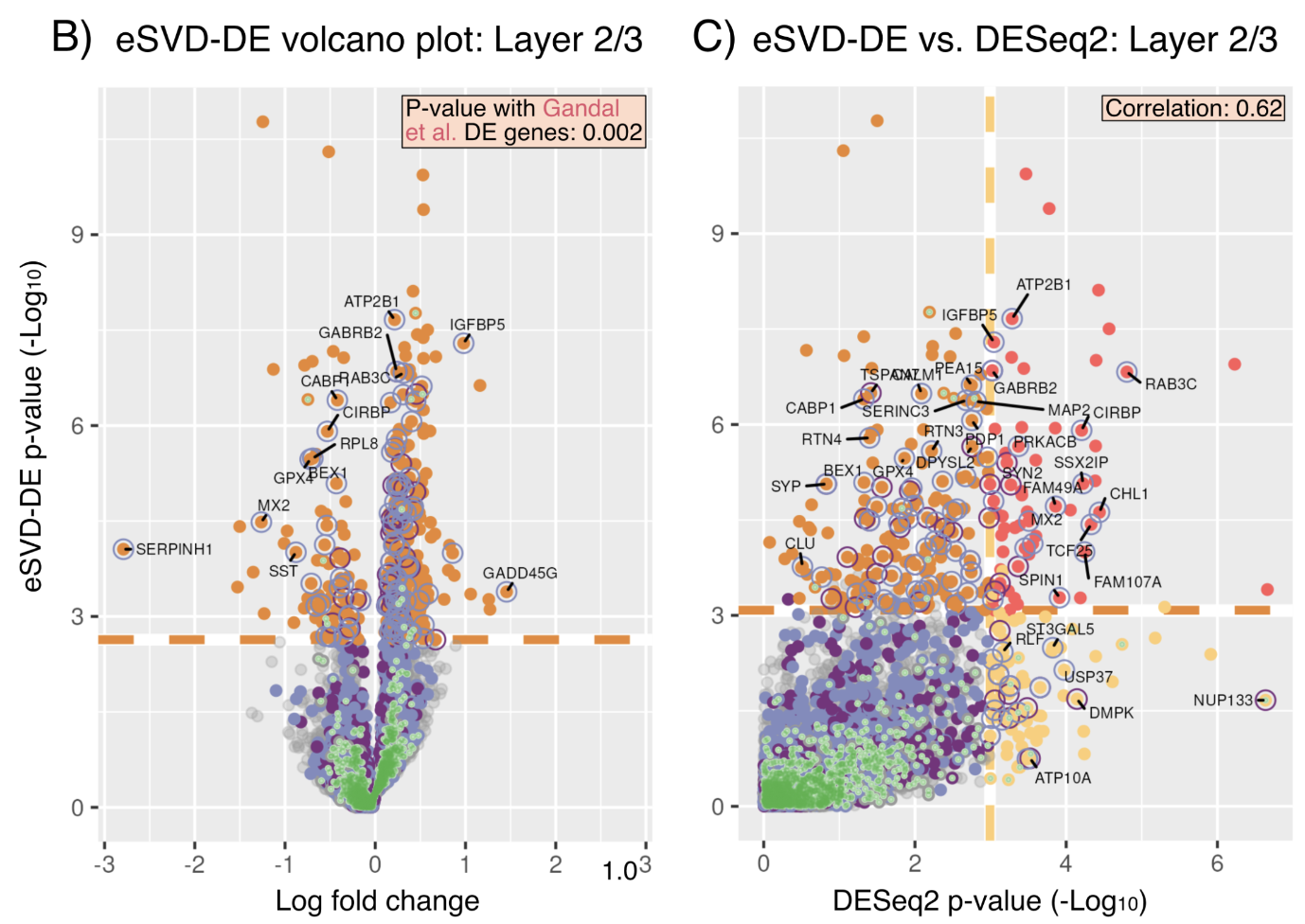
As single-cell RNA-seq (scRNA) transitions from studying cell-lines, clonal mice or a few individuals into studying cohorts of individuals, there is a need to create new differential expression (DE) methods to investigate DE among individuals instead of among cells. Towards this end, we tailor the eSVD, formerly a dimension-reduction tool, equipped with a downstream pipeline to perform DE in multi-individual scRNA data, where we both design our test statistics to account for variability among and within individuals as well as incorporating the individual-level covariates into the eSVD-DE framework. We show that this can recover reproducible signals across different datasets as well as achieves higher power thanks to its dimension-reduction framework.
eSVD-DE: Cohort-wide differential expression in single-cell RNA-seq data using exponential-family embeddings
BMC Bioinformatics 25.1 (2024): (link) (biorxiv) (code: method) (code: analysis) (code: tutorials)
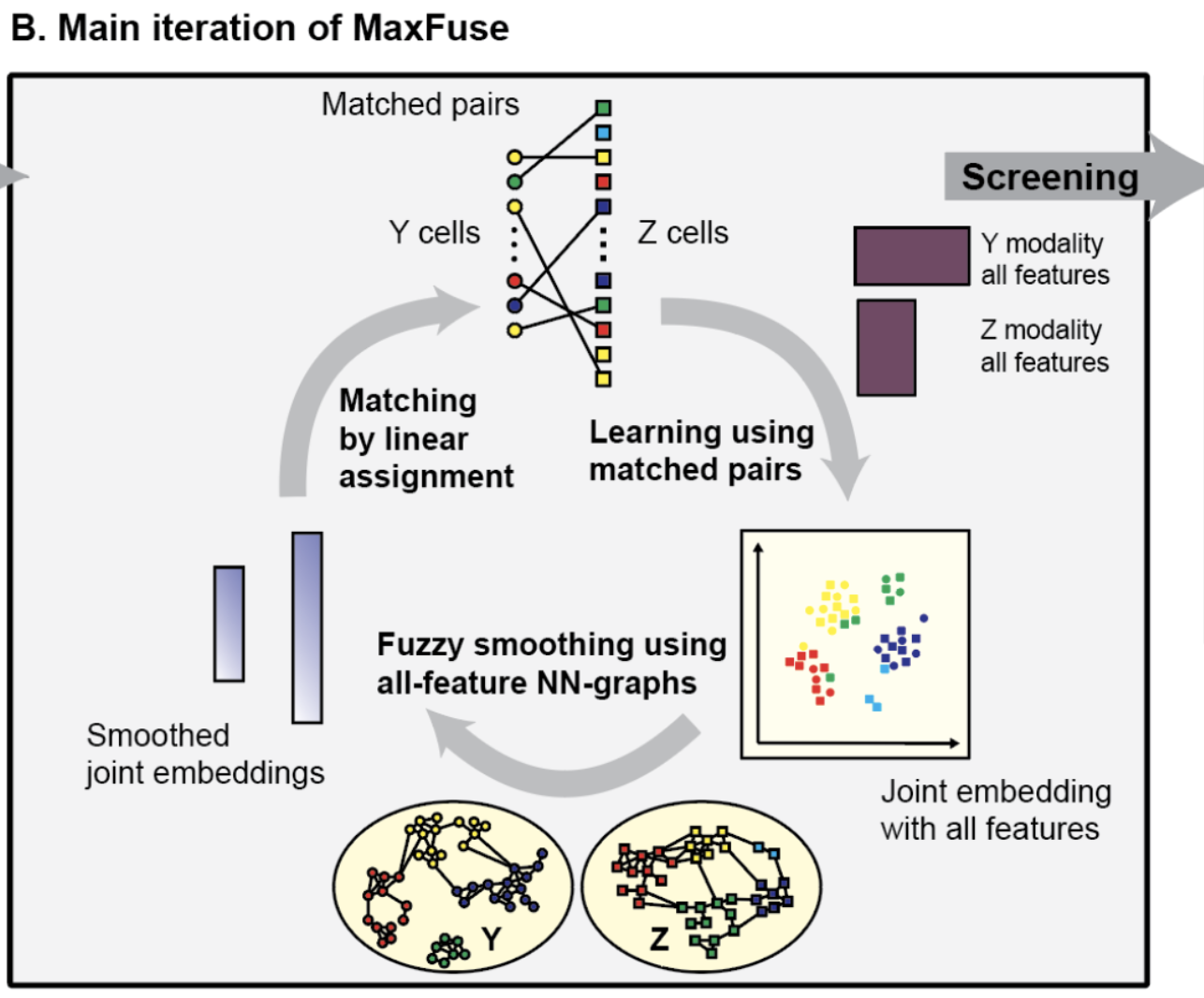
Different sequencing technologies offer complementary insights into a biological system, but it becomes difficult to integrate two datasets together when both datasets differ in the specific cells sequenced as well as which features are measured. Hence, using an iterative co-embedding based on CCA, data smoothing, and cell matching, we match cells in one dataset (i.e., one modality) to the other. The effectiveness of our method is demonstrated by integrating scRNA-seq and surface-antibody data, as well as scRNA-seq data with spatial proteomic data (such as CODEX) and scATAC-seq.
Integration of spatial and single-cell data across modalities with weak linkage
Nature Biotechnology 42 (2023): (link) (biorxiv) (code: method) (code: tutorials)
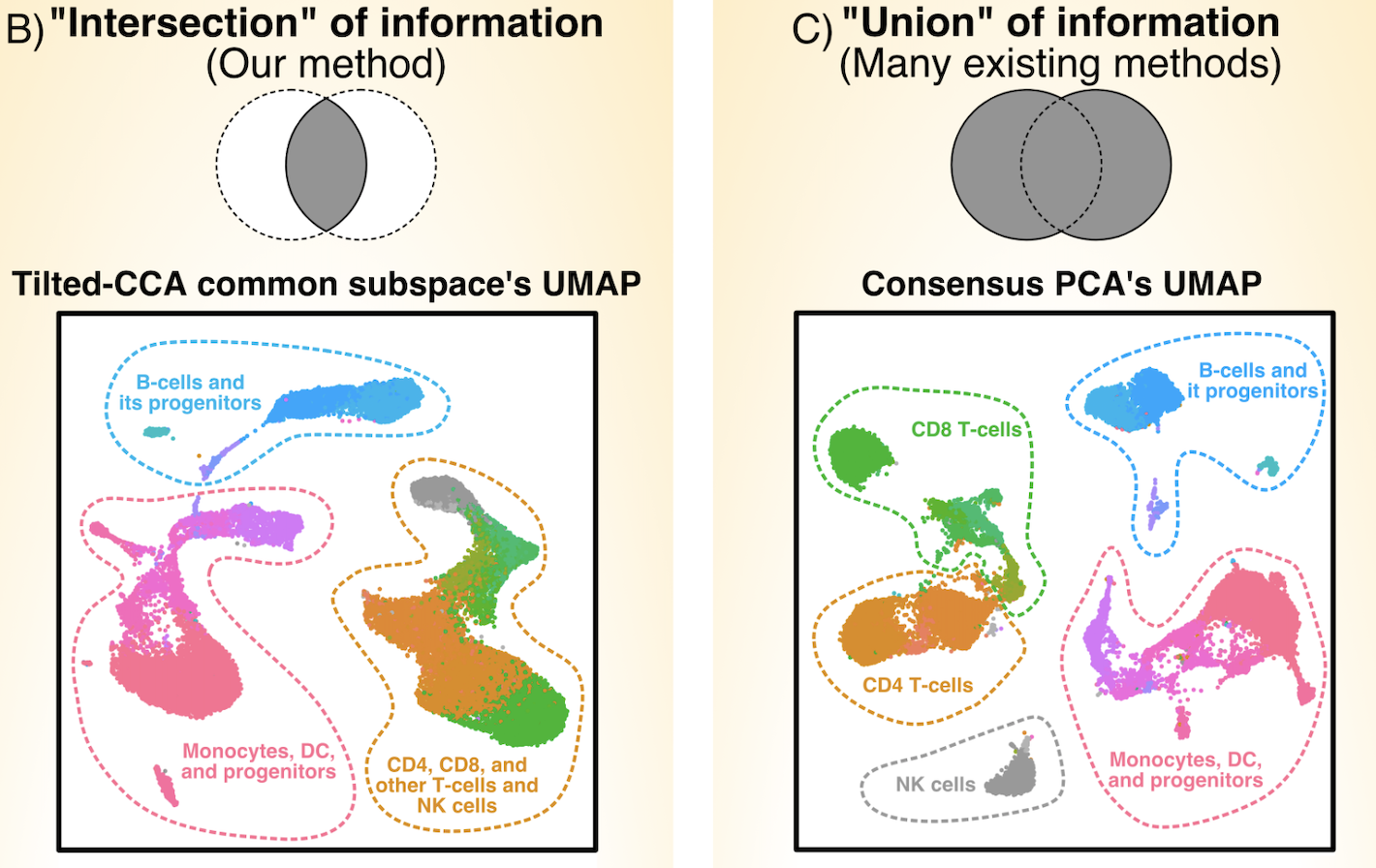
Paired multiomic single-cell data provides ample opportunities for biologists to study the relations between molecular modalities (such as gene expression and chromatin peaks), but it difficult to assess what kind of signals are shared in common between the two modalities. We developed the Tilted-CCA, which shows the "intersection of information" between the two modalities, in contrast with many existing dimension-reduction methods that show the "union of information." In particular, the matrix decomposition provided by Tilted-CCA allows us to design targeted antibody panels for RNA and surface-antibody technologies, as well as uncover the terminal cell-states as well as the relation between chromatin open/closed cell-states and gene expression in developmental systems.
Quantifying common and distinct information in single-cell multimodal data with Tilted-CCA
Proceedings of the National Academy of Sciences (PNAS) 120.32 (2023): (link) (biorxiv) (code: method) (code: analysis) (code: tutorials)
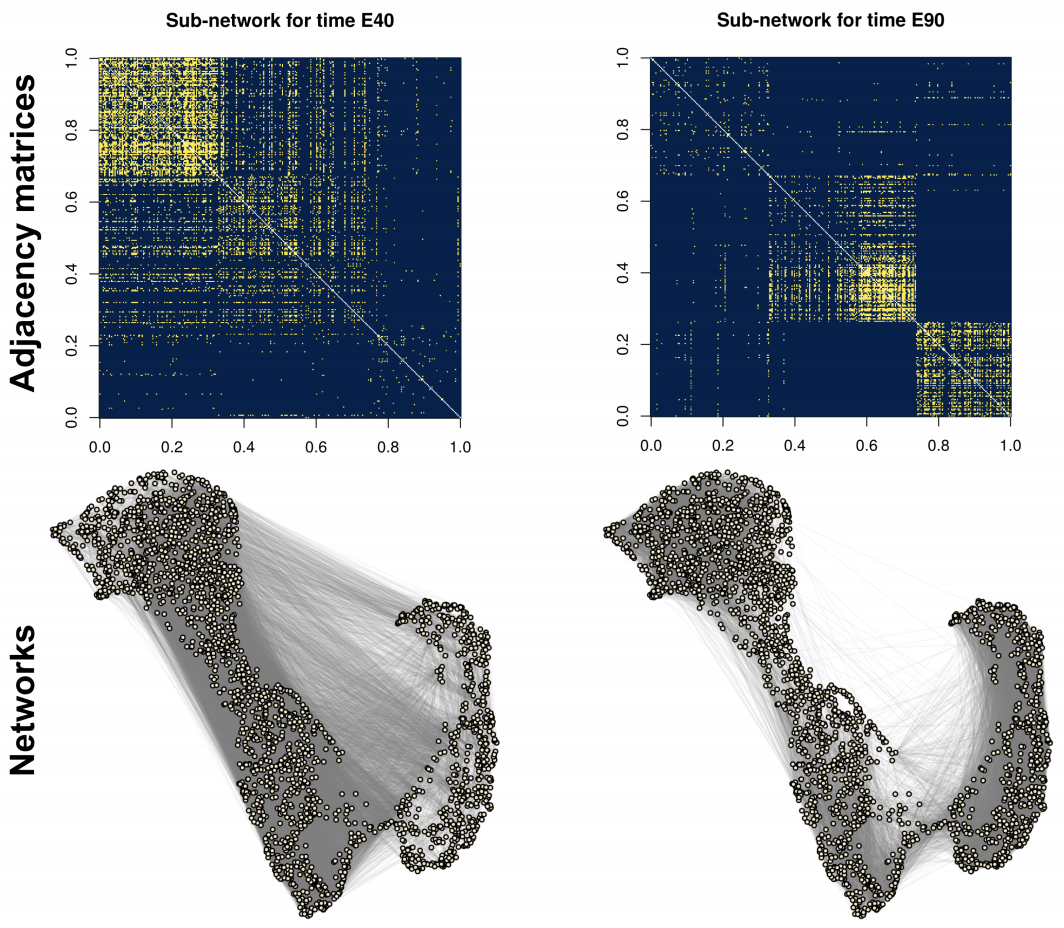
While clustering of nodes is now theoretically well-understood for a single network through the lense of spectral embeddings, the theoretical understanding for clustering in multi-layer networks is less well-understood. In this paper, when the clustering structure is shared across all the layers, we develop a simple way to aggregate information across layers via squaring the adjacency matrix with an appropriate bias-adjustment. Our theoretical analysis shows we can both allow for networks with dissociative structure (i.e., negative eigenvalues) and provably obtains consistent clustering even if each individual network is extremely sparse. We apply our method to study cluster of genes in developing monkey brains.
Bias-adjusted spectral clustering in multi-layer stochastic block models
Journal of the American Statistical Association (JASA) 118 (2023): (link) (arxiv) (git)
Data (4.15 Gb, Zip file): (dropbox link) (readme)
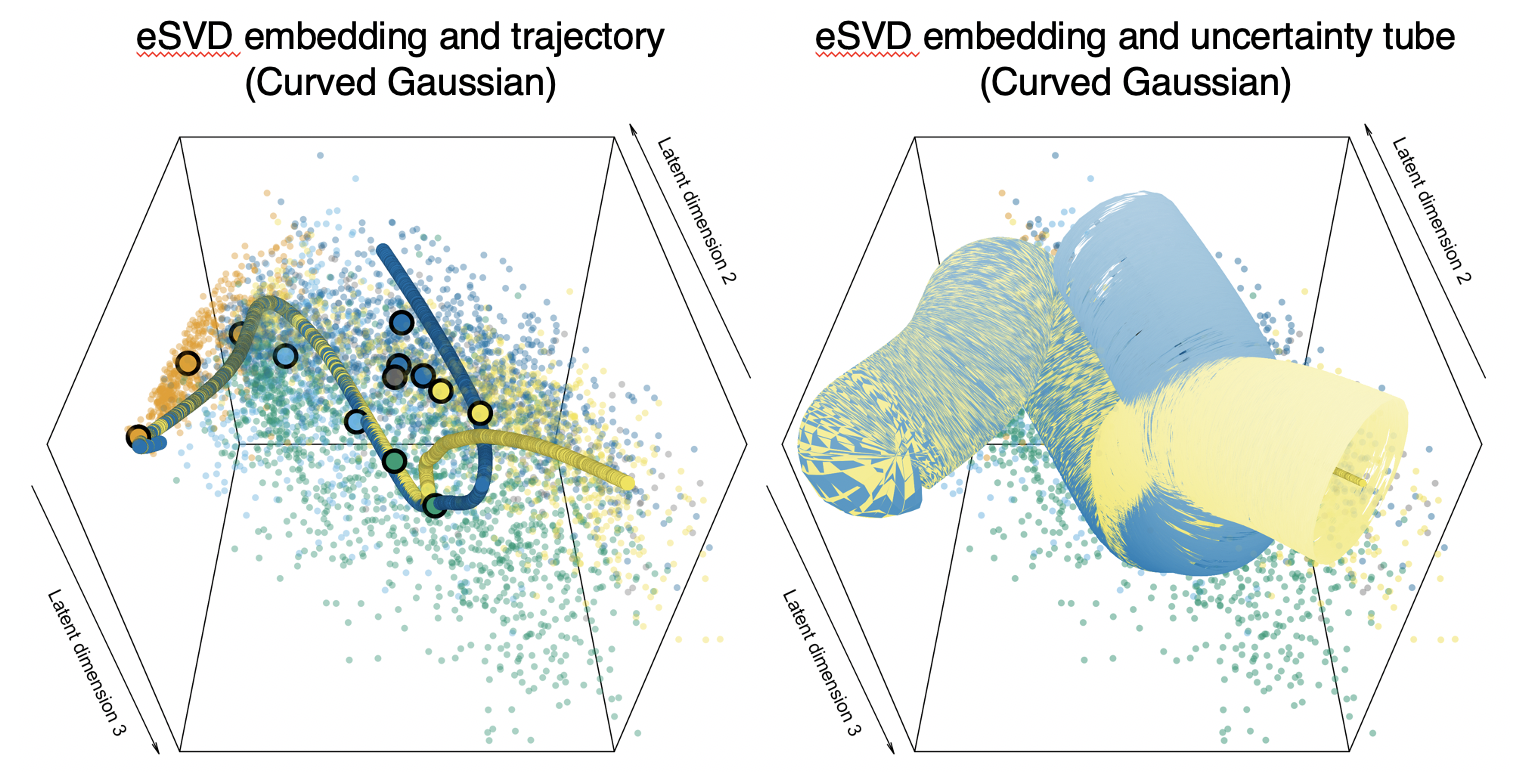
Common pipelines to estimate the cell developmental trajectories based on single-cell data typically first embed each cell into a lower-dimensional space, but these embedding typically assume statistical models that do not model single-cell data well. In this paper, we develop an embedding for hierarchical model where the inner product between two latent low-dimensional vectors is the natural parameter of an exponential family distributed random variable, and prove identifiability and convergence. When studying oligodendrocytes in fetal mouse brains, we find that oligodendrocytes mature into various cell types.
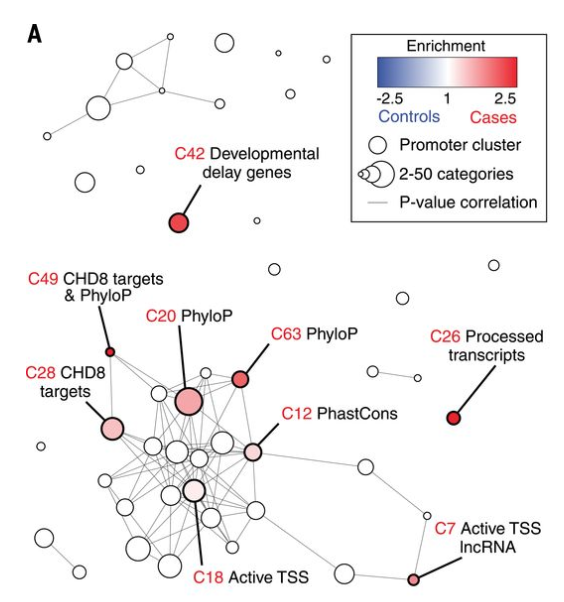
While de novo mutations within the protein-coding portion of the genome have been thoroughly studied, these mutations in the noncoding portions which comprise of 98.5% of the genome have been less well understood. In this paper, we use a bioinformatics framework that builds upon (among other things) sparse PCA and DAWN based on simulated null datasets to analyze 1902 autism quartets via WGS and find that the strongest signals arose from promoters -- noncoding regions that control gene transcription.
Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder
Science 362.6420 (2018): (link) (pdf) (git)
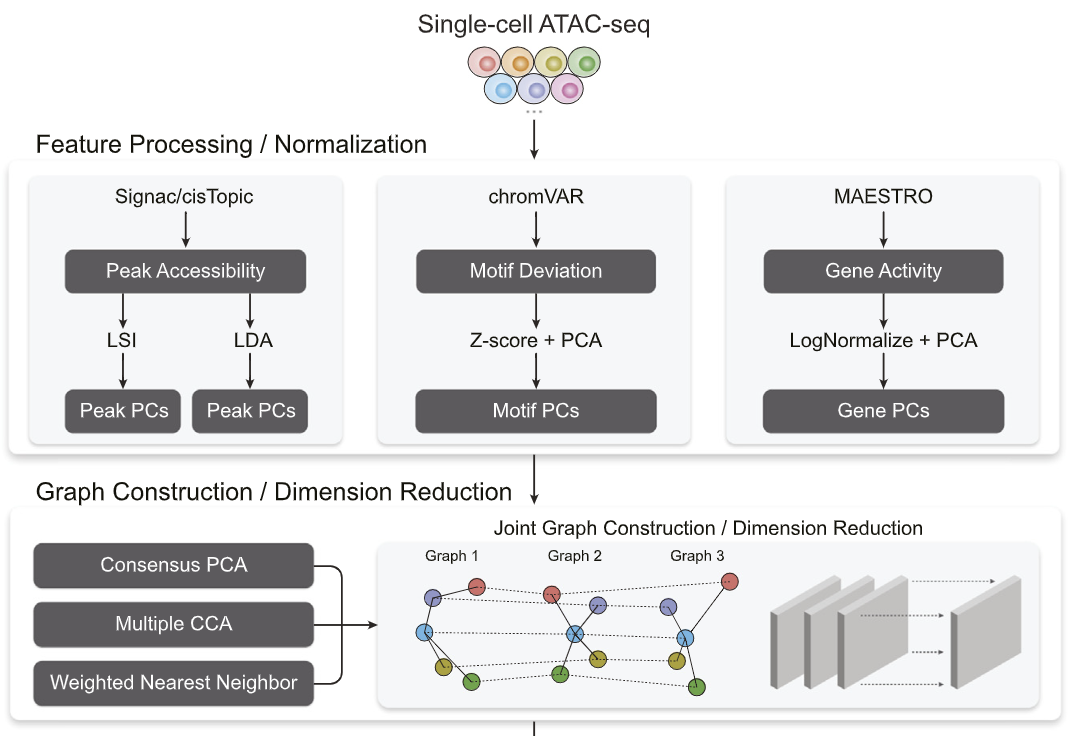
There are different signals a researcher can extract from single-cell ATAC-seq, and each has their own biological relevance for downstream analysis. However, one might want to perform computational tasks that aggregate all such signals together. We propose a computational framework to aggregate across such signals for downstream visualization, cell-clustering and trajectory inference.
Destin2: Integrative and cross-modality analysis of single-cell chromatin accessibility data
Frontiers in Genetics 14 (2023): (link) (biorxiv)
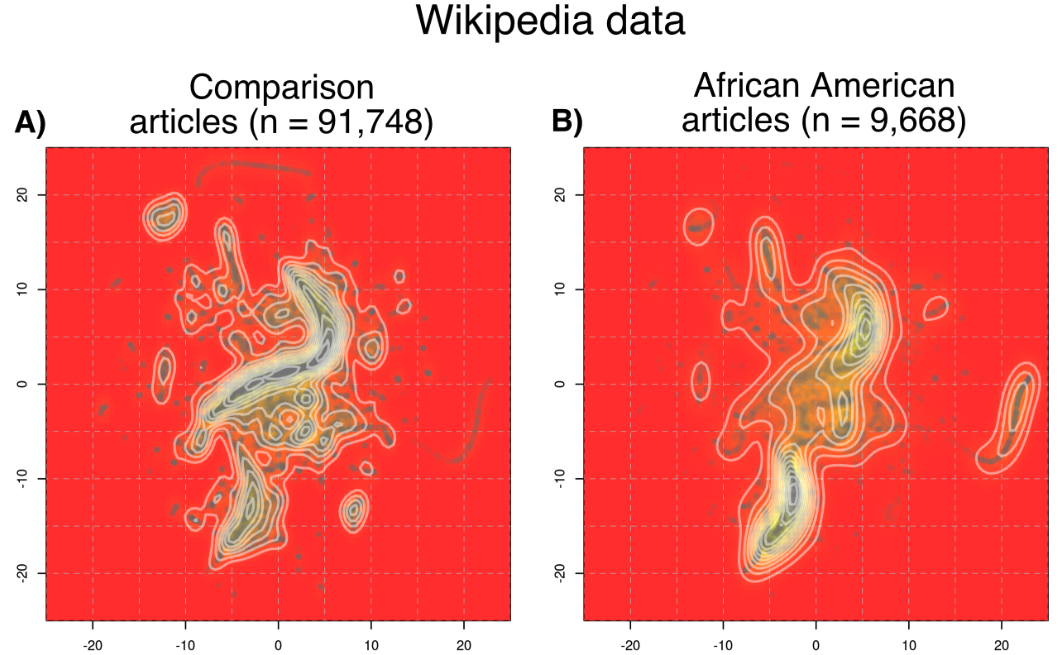
Social biases can influence Wikipedia articles of individuals across different genders and ethnicities that result in disproportiate article lengths, number of languages written about said individuals, etc., but these differences can be difficult to quantify appropriately. In this paper, we develop a matching method to find an appropriate comparison set of articles for each target group of interest (ex: articles of African American individuals) based on the articles' categories. We show the differing covariate distributions across the different target groups, and uncover quantitative results that reinforce existing social theories. This paper won the Wikimedia Foundation Research Award of the Year in 2023.
Controlled Analyses of Social Biases in Wikipedia Bios
Proceedings of the ACM Web Conference 2022 (2022): (link) (arxiv) (git)
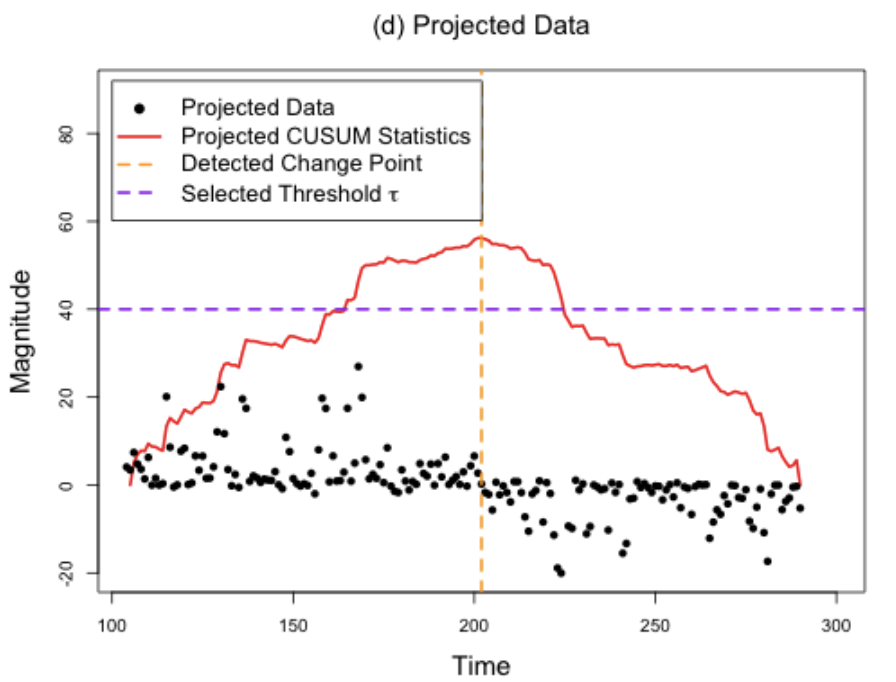
Methods to detect multiple changepoints in regression coefficients typically have localization rate (i.e., rate to estimate the location of the changepoints) that scale inversely with the square root with number of samples. In this paper, we develop a method that appropriately transforms the regression problem into a one-dimensional problem upon which we apply WBS. We prove the convergence rate scales inversely with number of samples, a substantial improvement over existing rates.
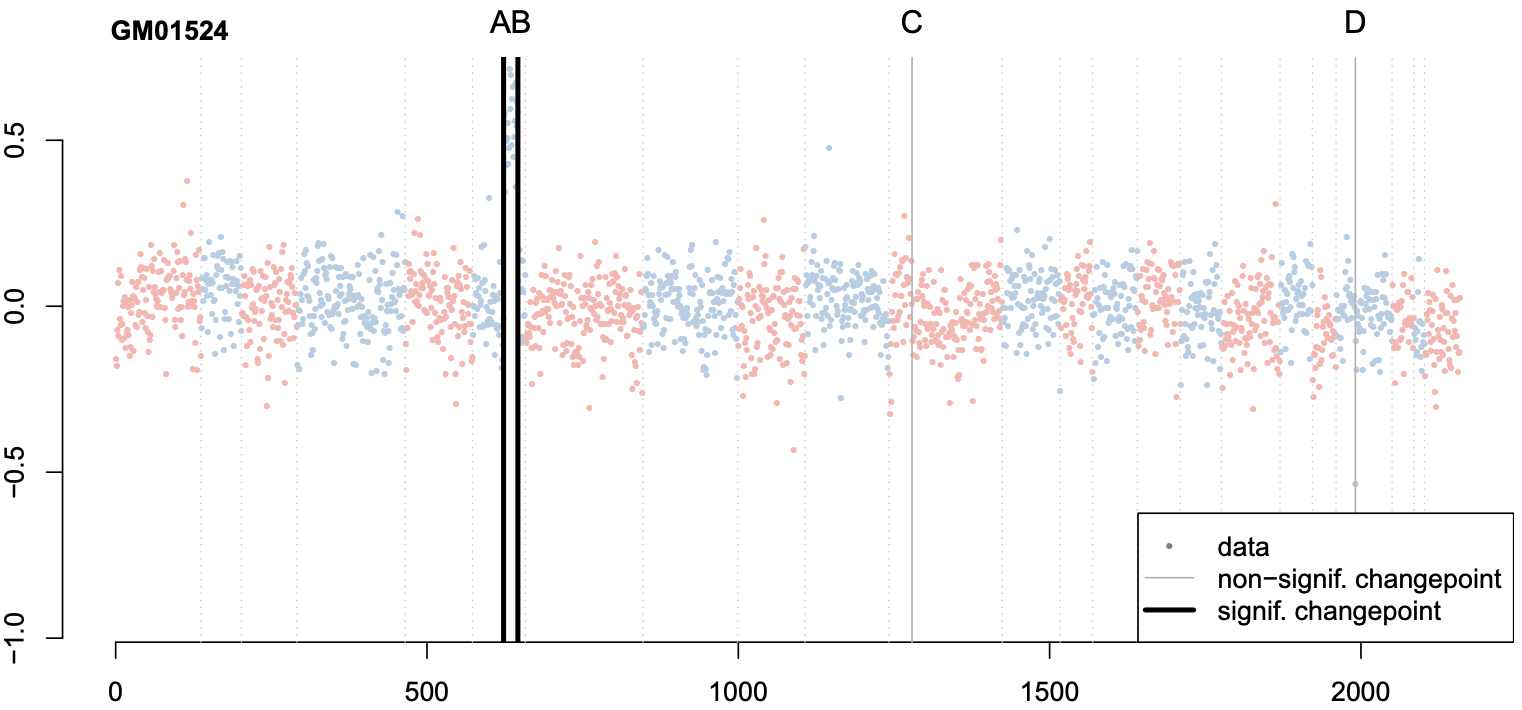
Changepoint detection methods such as binary segmentation are often used in CGH analyses for copy number variation detection, but these methods lack proper downstream statistical inference. In this paper, we develop post-selection hypothesis tests for various changepoint detection methods, prove our sampling strategies' validity, and provide substantial practical guidelines based on simulation.
Valid post-selection inference for segmentation methods with application to copy number variation data
Biometrics 77.3 (2021): 1037-1049 (link) (arxiv) (pdf) (git 1, git 2)
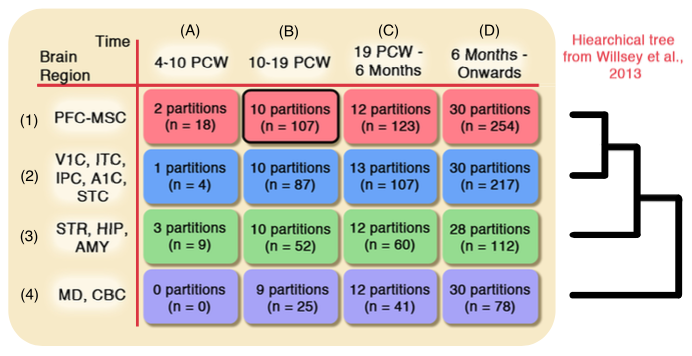
Microarray samples from brain tissue is hard to collect, and also varies substantially depending on the tissue's brain region and the developmental age of its subject, hence it is hard to collect enough samples for the statistical analysis. In this paper, we develop a sample selection method to find additional microarray samples that are statistically similar to the samples of our desired spatio-temporal brain tissue. We demonstrate that after apply an existing analysis pipeline to our selected samples, we detect a higher percentage of autism risk genes.
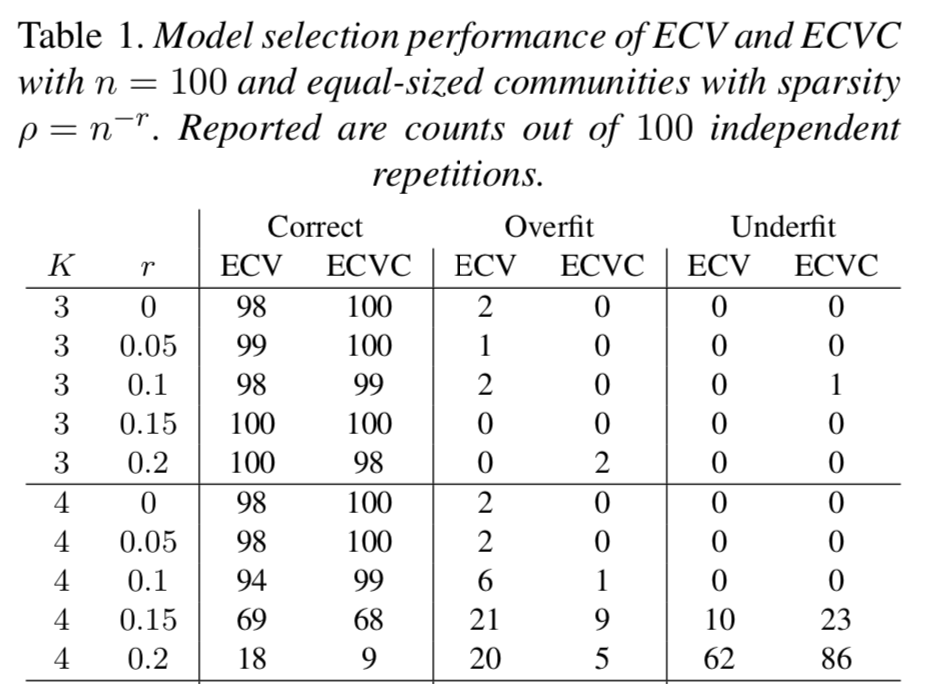
We discuss "Network cross-validation by edge sampling" by Li, Levina, and Zhu (Biometrika, 2020) where empirically explore the possibilities of combining the ideas of the authors with ideas providing confidence sets for cross-validated parameters as well as extending the ideas of the authors into the network tensor setting.
Discussion of 'Network cross-validation by edge sampling'
Biometrika 107.2 (2020): 285-287. (link) (pdf) (git)
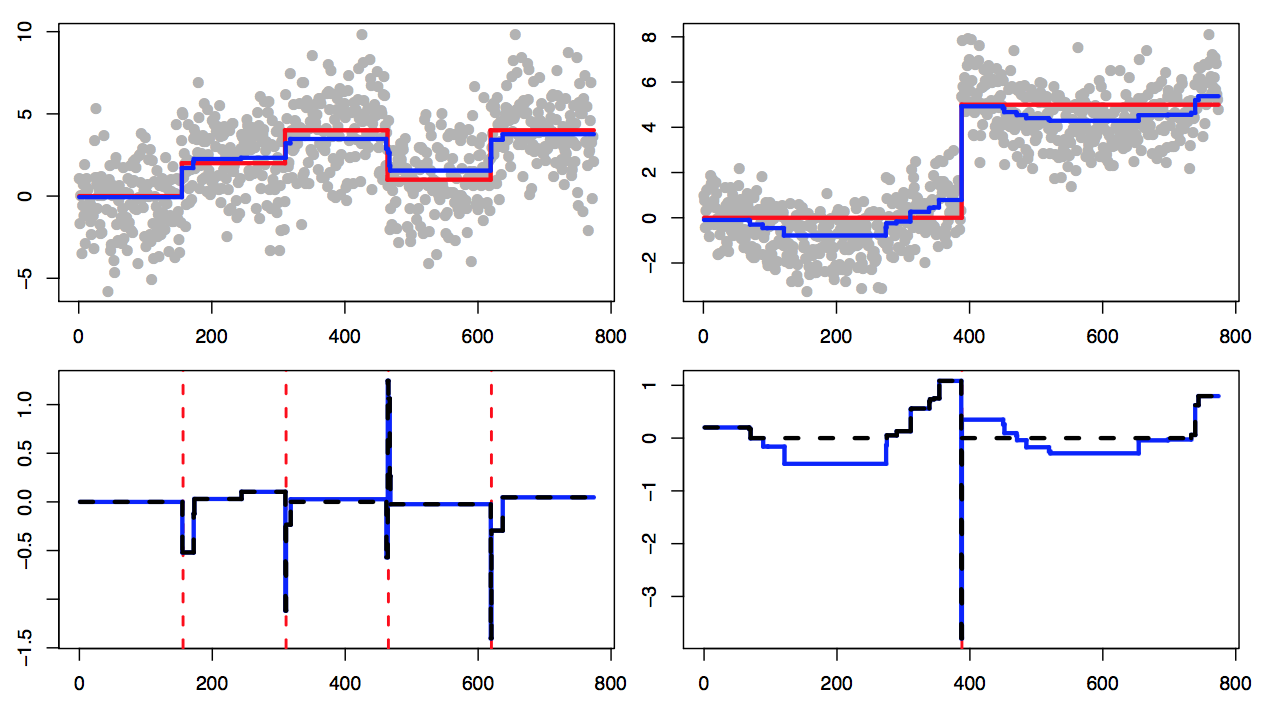
Changepoint estimators have statistical theory for how well they estimate the mean function and how well they estimate the changepoints, but existing theory often analyzes these properties separately. In this paper, we prove a near-optimal estimation rate for the fused lasso, which in turn directly proves a changepoint detection rate that is near the detection limit. We extend this logic to other estimators and settings.
A sharp error analysis for the fused Lasso, with application to approximate changepoint screening
Advances in Neural Information Processing Systems (NeurIPS) (2017): (link) (arxiv) (git)
Many compressed sensing are developed to be as generic as possible, but have shortcomings in specialized settings where modern optimization theory can deliver a substantial boost in computational efficiency. In this paper, we develop two compressed sensing algorithms, one specialized for extremely sparse signals and another specialized for Kronecker-structed sensing matrices. We numerically demonstrate a near 10-times reduction in computation time compared to other state-of-the-art methods.
Revisiting compressed sensing: Exploiting the efficiency of simplex and sparsification methods
Mathematical Programming Computation 8.3 (2016): 253-269. (link) (pdf) (git)
Last Updated: September 13, 2025